1 hætteglas (11,7 ml) indeholder 1400 mg rituximab (1 ml indeholder 120 mg rituximab).
| Navn | Pakkens indhold | Det aktive stof | Pris 100% | Sidst ændret |
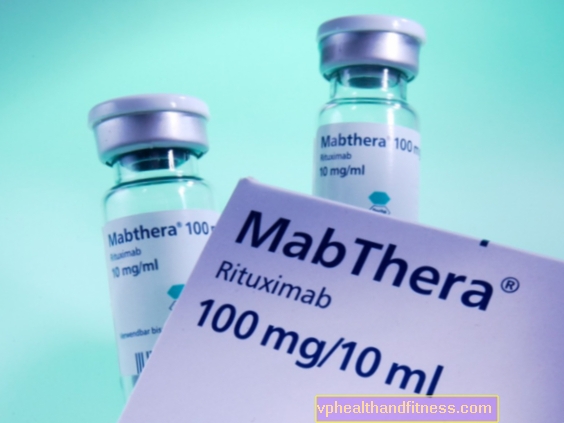
| MabThera® | 1 hætteglas 11,7 ml, sol. for chok subs. | Rituximab | 2019-04-05 |
Handling
Rituximab er et kimært humant-murint monoklonalt antistof produceret ved genteknologi. Det er et glykosyleret immunglobulin, der indeholder humane IgG1 konstante sekvenser og musesekvenser med let og tung kæde. Antistoffet produceres i en kinesisk hamster ovariecellesuspensionskultur og oprenses ved hjælp af selektiv kromatografi og ionbytningsmetoder og specifikke virusinaktiverings- og fjernelsesprocedurer. Rituximab binder specifikt til CD20-transmembranantigenet, som er et ikke-glycosyleret phosphoprotein, der findes på præ-B-lymfocytter og modne B-lymfocytter. Fab-domænet af rituximab binder til CD20-antigenet på B-lymfocytter og aktiverer immunsystemets lysemekanismer gennem Fc-domænet. B. Mulige mekanismer for cellelyse inkluderer komplementafhængig cytotoksicitet (CDC) associeret med bindingen af komponent C1q og antistofafhængig cellulær cytotoksicitet (ADCC) medieret af en eller flere typer Fcy-receptorer på overfladen af granulocytter, makrofager og NK-lymfocytter. Binding af rituximab til CD20-antigenet på B-lymfocytter har også vist sig at inducere celledød gennem apoptose. Efter subkutan administration absorberes rituximab langsomt og når C efter ca. 3 dage. Den absolutte biotilgængelighed er ca. 71%. Rituximab-eksponering øger dosis proportionalt over dosisområdet 375 mg / m2 til 800 mg / m2 (subkutan administration). De farmakokinetiske parametre (clearance, distributionsvolumen og eliminationshalveringstid) er ens for den subkutane formulering og den intravenøse formulering. Den estimerede mediane eliminering af T0.5 for subkutant administreret lægemiddel er 29,7 dage (interval 9,9 til 91,2 dage). Lægemidlet til subkutan administration indeholder rekombinant human hyaluronidase (rHuPH20), et enzym, der anvendes til at øge dispersionen og absorptionen af samtidigt administrerede lægemidler i tilfælde af subkutan injektion.
Dosering
Subkutant. Voksne: 1400 mg uanset patientens kropsoverfladeareal. Før rituximab-opløsning til injektion under huden påbegyndes, skal alle patienter først få den fulde dosis rituximab-opløsning til infusion intravenøst. Patienter, der ikke har været i stand til at få en fuld dosis intravenøs infusion, bør få rituximab-infusion til efterfølgende behandlingscyklusser, indtil den fulde intravenøse dosis er blevet administreret. Skift til den subkutane form af lægemidlet er kun mulig i den anden eller efterfølgende behandlingscyklus. Follikulære ikke-Hodgkin lymfomer. Kombinationsterapi. Dosen af rituximab i kombination med kemoterapi til induktionsbehandling af tidligere ubehandlede patienter med follikulært non-Hodgkins lymfom eller hos patienter med tilbagefald eller ildfast follikulært lymfom: i den første cyklus, rituximab som en opløsning til infusion i en dosis på 375 mg / m2, efterfulgt af rituximab-opløsning til injektion under huden i en fast dosis på 1400 mg / cyklus i op til 8 på hinanden følgende cyklusser. Rituximab bør administreres på dag 1 i hver kemoterapicyklus efter intravenøs administration af glukokortikoidkomponenten i kemoterapien, hvor det er relevant. Støttende pleje. Tidligere ubehandlet follikulært ikke-Hodgkins lymfom: Den anbefalede vedligeholdelsesdosis af rituximab subkutan injektionsopløsning til tidligere ubehandlet follikulær ikke-Hodgkins lymfomresponder er 1.400 mg hver anden måned (2 måneders start fra den sidste dosis af induktionsterapi) indtil sygdomsprogression eller i en maksimal periode på 2 år (i alt 12 infusioner). Patienter med recidiverende eller ildfast follikulært lymfom: Den anbefalede dosis af rituximab subkutan injektionsopløsning til vedligeholdelsesbehandling hos patienter med recidiverende eller refraktær follikulært lymfom, som reagerede på induktionsterapi, er: 1.400 mg hver 3. måned (startende 3 måneder efter den sidste dosis af induktionsterapi) indtil sygdomsprogression eller i en maksimal periode på 2 år (i alt 8 infusioner). Diffuse store B-celle-ikke-Hodgkins lymfomer Rituximab bør anvendes i kombination med kemoterapi i henhold til CHOP-regimen (cyclophosphamid, doxorubicin, vincristin, prednisolon). I den første cyklus gives rituximab som en intravenøs infusion på 375 mg / m2 efterfulgt af en konstant dosis på 1400 mg / cyklus subkutan injektionsvæske, opløsning til i alt 8 cyklusser. Rituximab bør administreres på dag 1 i hver kemoterapicyklus efter forudgående intravenøs administration af glukokortikoidkomponenten i CHOP-regimet. Sikkerheden og effekten af rituximab i kombination med andre kemoterapiregimer ved behandling af diffust stort B-celle-non-Hodgkins lymfom er ikke fastlagt. Dosisændringer under behandlingen. Ingen dosisreduktion af rituximab anbefales. Standarddosisreduktioner for kemoterapeutiske midler bør anvendes, når lægemidlet anvendes i kombination med kemoterapi. Særlige patientgrupper. Dosisjustering er ikke nødvendig hos ældre patienter (> 65 år). Sikkerheden og effekten af lægemidlet hos børn og unge er endnu ikke fastlagt. Lægemidlet skal administreres under nøje overvågning af en erfaren læge et sted, hvor alle de nødvendige ressourcer til genoplivning er umiddelbart tilgængelige. Patienter skal observeres i mindst 15 minutter efter indgift af lægemidlet og længere hos patienter med højere risiko for overfølsomhedsreaktioner. Antipyretisk og antihistamin-præmedicinering (f.eks. Paracetamol og diphenhydramin) bør administreres inden hver administration af rituximab. Forbehandling med et glukokortikoid bør overvejes hos patienter med ikke-Hodgkins lymfom, som ikke får rituximab i kombination med glukokortikoidholdig kemoterapi. Det er vigtigt at kontrollere etiketten på lægemidlet hver gang for at sikre, at du får den rigtige medicin (intravenøs eller subkutan) og dosis til dig. Rituximab-opløsning til subkutan injektion bør kun administreres ved subkutan injektion i ca. 5 minutter. Injektionsnålen skal placeres på sprøjten umiddelbart før administration for at forhindre tilstopning. Injicér kun subkutant i underlivet (ingen data er tilgængelige til injektion andetsteds i kroppen). Brug aldrig på områder med rødme, ømhed eller hærdning af huden, blå mærker, modermærker eller ar. Under behandling med præparatet administreres andre lægemidler administreret subkutant bedst på forskellige steder. Hvis injektionen afbrydes, kan den genstartes på det samme eller et andet sted, hvis det er relevant.
Indikationer
Behandling af ikke-Hodgkins lymfom (NHL) hos voksne: behandling af tidligere ubehandlede patienter med fase III-IV follikulært lymfom i kombination med kemoterapi; vedligeholdelsesbehandling af patienter med follikulært non-Hodgkins lymfom, som har reageret på induktionsterapi; behandling af patienter med diffuse store B-celle-non-Hodgkins lymfomer med et positivt CD20-antigen i kombination med kemoterapi i henhold til CHOP-regimen (cyclophosphamid, doxorubicin, vincristin, prednisolon).
Kontraindikationer
Overfølsomhed over for rituximab, hyaluronidase, enhver anden ingrediens i præparatet eller over for museproteiner. Aktive, alvorlige infektioner. Patienter i en alvorligt immunkompromitteret tilstand.
Forholdsregler
For at forbedre sporbarheden af biologiske præparater skal handelsnavnet på det administrerede lægemiddel og batchnummeret tydeligt angives (eller cirkuleres) i patientens fil. Brug af lægemidlet som monoterapi hos patienter med klinisk stadium III-IV follikulært lymfom, ildfast kemoterapi eller i det andet eller efterfølgende tilbagefald efter kemoterapi, bør ikke anbefales, da sikkerheden ved rituximab-opløsning til subkutan injektion ikke er fastlagt. En gang om ugen På grund af den øgede risiko for progressiv multifokal leukoencefalopati (PML) skal patienter, der bruger lægemidlet, overvåges regelmæssigt for neurologiske symptomer eller forekomsten af symptomer, der tyder på PML. Hvis der er mistanke om PML, bør behandlingen afbrydes straks, indtil en diagnose er udelukket. Hvis der er nogen diagnostiske tvivl, skal MR udføres med kontrast, undersøgelse af cerebrospinalvæske skal udføres for at bestemme JC-virus-DNA, og neurologisk revurdering skal udføres. Vær især forsigtig, da symptomerne på PML kan gå ubemærket hen for patienten. Hvis en patient udvikler symptomer på PML, skal behandlingen med præparatet seponeres permanent. Behandling med præparatet er forbundet med forekomsten af lægemiddelrelaterede reaktioner, som kan være forbundet med frigivelse af cytokiner og / eller andre kemiske mediatorer. Cytokinfrigivelsessyndrom, tumorlysesyndrom og anafylaktiske reaktioner og overfølsomhedsreaktioner er ikke relateret til administrationsvejen og forekommer ved både intravenøs og subkutan rituximab-behandling. Alle patienter skal først få den fulde dosis af intravenøs rituximab, før de begynder subkutan administration. Den største risiko for lægemiddelrelaterede reaktioner observeres generelt i den første behandlingscyklus, og derfor vil initiering af behandling med intravenøs rituximab give en mere effektiv lindring af bivirkninger ved at bremse eller afbryde infusionen. Alvorligt cytokinfrigivelsessyndrom forekommer oftest inden for de første eller første 2 timer efter start af den første infusion. Præparatet skal anvendes med særlig forsigtighed hos patienter med en stor tumorbyrde eller med et stort antal (≥25 x 109 / l) cirkulerende tumorceller på grund af den øgede risiko for alvorligt cytokinfrigivelsessyndrom. Overvej at reducere den første infusionshastighed hos disse patienter eller dele dosis over 2 dage i cyklus 1 og hver efterfølgende cyklus, hvis lymfocytantal stadig er> 25 x 109 / L. Patienter med respirationssvigt eller tumorinduceret lungeinfiltration har en særlig risiko for komplikationer og bør behandles med øget forsigtighed. Hos patienter, der udvikler et alvorligt cytokinfrigivelsessyndrom, skal infusionen seponeres straks og intensiv symptomatisk behandling indledes. Da klinisk forringelse kan forekomme efter en indledende forbedring, bør alle patienter overvåges nøje, indtil tumorlysesyndrom og lungeinfiltration er udelukket.Efter intravenøs administration af proteinprodukter kan anafylaktiske eller andre overfølsomhedsreaktioner også forekomme (i modsætning til cytokinfrigivelsessyndrom forekommer de normalt inden for de første minutter efter infusionen) lægemidler til behandling af overfølsomhedsreaktioner skal være tilgængelige til øjeblikkelig brug under rituximab-administration. De resterende reaktioner, rapporteret i nogle få tilfælde, inkluderede hjerteinfarkt, atrieflimren, lungeødem og akut reversibel trombocytopeni. På grund af den mulige forekomst af hypotension under infusionen, bør det overvejes at trække de antihypertensive lægemidler tilbage 12 timer før rituximab-infusionen. Subkutan administration har været forbundet med udviklingen af milde eller moderate (grad 1 eller 2) hudreaktioner, som normalt forsvandt uden specifik behandling. Behandling med rituximab bør seponeres permanent i tilfælde af alvorlige hudreaktioner såsom Stevens-Johnsons syndrom eller toksisk epidermal nekrolyse. Det bør anvendes med forsigtighed til patienter med et neutrofiltal på 9 / l og / eller et antal blodplader på 9 / l på grund af den begrænsede kliniske erfaring hos sådanne patienter. Komplet blodtal, inklusive antal neutrofiler og blodplader, bør overvåges regelmæssigt under behandlingen. Der skal udvises særlig forsigtighed, når man overvejer at anvende præparatet til patienter med tidligere tilbagevendende eller kroniske infektioner og hos patienter med underliggende sygdomme, som kan disponere patienter for svære infektioner. Anvendelsen af rituximab kan være forbundet med hepatitis B-reaktivering (med risiko for fulminant hepatitis med dødelig udgang) hos både HBsAg + ve (HBsAg + ve) og HBsAg + ve-negative patienter. , men et anti-HB-kerneantigen-antistof (HBsAg-ve / HBcAb + ve) er blevet påvist, især når lægemidlet gives i kombination med glukokortikoider eller kemoterapi. Hepatitis B-screening, herunder mindst HBsAg- og HBcAb-test, skal udføres hos alle patienter, inden behandling med rituximab påbegyndes. Diagnostik bør suppleres med vurdering af andre infektionsmarkører i overensstemmelse med lokale anbefalinger. Patienter med aktiv hepatitis B-infektion bør ikke behandles med rituximab. Patienter, der er serologisk positive over for HBV - HBsAg og / eller HBcAb-infektion (men ingen kendt aktiv sygdomsstatus), inden de begynder med rituximab-behandling, skal konsulteres af en specialist i infektionssygdomme, før de overvåges og nøje overvåges i henhold til lokale standarder. for at forhindre reaktivering af hepatitis B. På grund af den øgede risiko for kardiotoksicitet bør patienter med en hjertesygdom i anamnesen og dem, der har modtaget kardiotoksisk kemoterapi, overvåges nøje.
Uønsket aktivitet
Sikkerhedsprofilen for rituximab-opløsning til subkutan injektion svarer til den, der ses med intravenøs formulering, bortset fra lokale hudreaktioner. Lokale hudreaktioner er meget almindelige hos patienter, der får rituximab subkutant. De mest almindelige lokale hudreaktioner er erytem (13%), smerter på injektionsstedet (7%) og hævelse på injektionsstedet (4%). Bivirkninger efter subkutan administration er milde til moderate. Alvorlige subkutane lægemiddelreaktioner (grad ≥3) kan omfatte grad 3 udslæt på injektionsstedet og mundtørhed. Lokale hudreaktioner af enhver kvalitet forekommer hyppigst i den første cyklus, derefter den anden cyklus, og falder med hver efterfølgende injektion. Følgende er bivirkninger, der er rapporteret ved brug af intravenøs rituximab hos patienter med non-Hodgkins lymfom (NHL) og kronisk lymfocytisk leukæmi (CLL), enten alene eller i kombination med kemoterapi. De mest almindeligt rapporterede alvorlige bivirkninger var infusionsrelaterede reaktioner (inklusive cytokinfrigivelsessyndrom, tumorlysesyndrom), infektioner og kardiovaskulære hændelser. Andre rapporterede alvorlige bivirkninger var: alvorlige virusinfektioner, herunder infektioner forårsaget af herpesvirus (cytomegalovirus, varicella-zoster-virus og herpesvirus), hepatitis C-virus, hepatitis B-virus, JC-virus (Progressiv multifokal leukoencefalopati - PML) - Der er rapporteret om dødsfald. Hos patienter med allerede eksisterende Kaposis sarkom behandlet med rituximab er der set tumorprogression hos patienter med ikke-godkendte indikationer, og størstedelen af patienterne var HIV-positive. Meget almindelig: bakteriel infektion, virusinfektion, bronkitis, neutropeni, leukopeni, febril neutropeni, trombocytopeni, infusionsrelaterede bivirkninger, angioødem, kvalme, pruritus, udslæt, alopeci, pyreksi, kulderystelser, asteni, hovedpine, nedsat blodniveau IgG. Almindelig: sepsis, lungebetændelse, feberinfektion, helvedesild, luftvejsinfektioner, svampeinfektioner, infektioner med ukendt etiologi, akut bronkitis, bihulebetændelse, hepatitis B (inklusive primære infektioner og hepatitis B-reaktivering, mere almindelig hos patienter rituximab i kombination med cytotoksisk kemoterapi), anæmi, aplastisk anæmi, granulocytopeni, overfølsomhed, hyperglykæmi, vægttab, perifert ødem, ansigtsødem, stigning i LDH, hypokalcæmi, paræstesi, hypæstesi, agitation, søvnløshed, hovedpine, vasodilatation, svimmelhed angst, tåresygdom, konjunktivitis, tinnitus, øre smerter, myokardieinfarkt (herunder sjældent rapporterede tilfælde med dødelig udgang), arytmi, atrieflimren, takykardi, hjertelidelser, hypertension, ortostatisk hypotension, lavt blodtryk, bronkospasme (inklusive sjælden rapporterede dødelige tilfælde), luftvejssygdomme, smerter l bryst, dyspnø, øget hoste, rhinitis, opkastning, diarré, mavesmerter, dysfagi, stomatitis, forstoppelse, fordøjelsesbesvær, anoreksi, halsirritation, nældefeber, svedtendens, nattesved, hudlidelser, muskelspændinger, smerter muskelsmerter, artralgi, rygsmerter, nakkesmerter, smerter, tumorsmerter, rødme, utilpashed, forkølelsessyndrom, træthed, kulderystelser, multiorgansvigt (inklusive sjældne tilfælde med dødelig udgang). Ikke almindelig: koagulationsforstyrrelser, aplastisk anæmi, hæmolytisk anæmi, lymfadenopati, depression, nervøsitet, dysgeusi, venstre ventrikulær svigt, supraventrikulær takykardi, ventrikulær takykardi, angina pectoris, myokardisk iskæmi, bradykardi, astma, bronchiolitis obliteration, forstørret mave, smerter på injektionsstedet. Sjælden: alvorlige virusinfektioner, anafylaksi, alvorlige hjertehændelser (herunder sjældent rapporterede dødelige tilfælde), interstitiel lungesygdom (inklusive dødelige tilfælde). Meget sjælden: Forbigående stigning i serum IgM, tumorlysesyndrom og cytokinfrigivelsessyndrom (inklusive sjældent rapporterede dødelige tilfælde), serumsygdom, perifer neuropati, ansigtsnerveseparese, alvorligt synstab, hjertesvigt (herunder sjældent rapporterede tilfælde med dødelig), vaskulitis (overvejende kutan), leukocytoklastisk vaskulitis, respirationssvigt (herunder sjældent rapporterede fatale tilfælde), gastrisk eller tarmperforering (inklusive fatale tilfælde), svære blisterlignende hudreaktioner (Stevens-Johnsons syndrom og toksisk epidermal nekrolyse, inklusive dødelige tilfælde), nyresvigt (herunder sjældent rapporterede tilfælde med dødelig udgang). Ikke kendt: sen neutropeni, akut reversibel infusionsrelateret trombocytopeni, kranial neuropati, tab af andre sanser, høretab, lungeinfiltrater. Posterior reversibel encefalopatisyndrom (PRES) er rapporteret (forekom hos patienter med kendte risikofaktorer for PRES, herunder underliggende sygdom, hypertension, immunsuppressiv behandling og / eller kemoterapi).
Graviditet og amning
Lægemidlet bør ikke anvendes til gravide kvinder, undtagen i tilfælde, hvor de mulige fordele opvejer den potentielle risiko (der er en risiko for en reduktion i antallet af B-lymfocytter og udviklingen af lymfocytopeni hos nyfødte). Kvinder i den fødedygtige alder skal bruge effektiv prævention både under behandlingen og i 12 måneder efter behandlingen. Du bør ikke amme under behandlingen med præparatet og i 12 måneder efter afslutningen.
Kommentarer
Lægemidlet påvirker ikke evnen til at føre motorkøretøj eller betjene maskiner. Præparatet skal opbevares ved 2-8 ° C. Oplysninger udarbejdet på basis af SPC af 04/26/2018 Den nuværende produktresumé er tilgængelig på www.roche.pl.
Interaktioner
I øjeblikket er der begrænsede data om lægemiddelinteraktioner rituximab. Rituximab påvirkede ikke farmakokinetikken for fludarabin eller cyclophosphamid. Der var heller ingen effekt af fludarabin eller cyclophosphamid på rituximabs farmakokinetik. Patienter med humane anti-murine eller antikimære antistoffer (HAMA / HACA) kan udvikle allergiske eller overfølsomhedsreaktioner efter administration af andre monoklonale antistoffer til diagnostiske eller terapeutiske formål.
Præparatet indeholder stoffet: Rituximab
Refunderet stof: NEJ








-wartoci-odywcze.jpg)




-po-zabiegach-kardiologicznych.jpg)