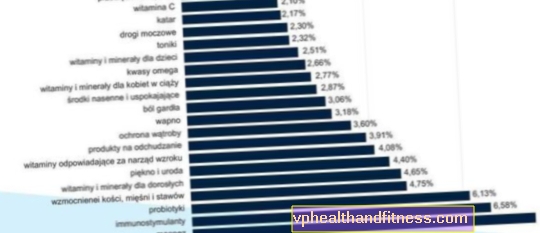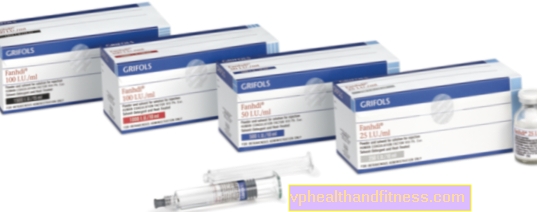
1 hætteglas indeholder 250 IE, 500 IE eller 1000 IE human blodkoagulationsfaktor VIII (FVIII) og henholdsvis 300 IE, 600 IE eller 1.200 IE den menneskelige von Willebrand-faktor. Den specifikke aktivitet er mindst 2,5 IE. op til 10 IE FVIII: C / mg protein afhængigt af pakningsstørrelsen (250 IE, 500 IE og 1000 IE). Produceret fra plasmaet fra menneskelige donorer.
| Navn | Pakkens indhold | Det aktive stof | Pris 100% | Sidst ændret |
| FANHDI | 1 hætteglas + 1 ampere sprøjte, pulver og rekonstitution til klargøring løsning for chok og / eller inf. | Faktor VIII, Faktor von Willebrand | 2019-04-05 |
Handling
Kombination af koagulationsfaktorer - faktor VIII og von Willebrand-faktor er fysiologiske komponenter i humant plasma, der fungerer som endogene komponenter. Administration af von Willebrand-faktor muliggør korrektion af hæmostatiske lidelser, der forekommer hos patienter med von Willebrand-faktormangel på to niveauer: von Willebrand-faktor genopretter vedhæftningen af blodplader til det vaskulære endotel på stedet for vaskulær skade (den fæstner sig til både endotelet og membranen af blodplader), hvilket resulterer i primær hæmostase, som manifesteres ved en reduktion i blødningstid (effekten vises straks og er stort set afhængig af niveauet af proteinpolymerisation); von Willebrand-faktor forårsager en forsinket stigning i samtidig faktor VIII-mangel, en intravenøs injektion af von Willebrand-faktor binder til endogen faktor VIII (som syntetiseres i patienten), stabilisering af denne faktor forhindrer dens hurtige nedbrydning. Af denne grund gendanner administration af ren von Willebrand-faktor (en lav faktor VIII von Willebrand-faktorpræparation) faktor VIII-niveauer til normal som en sekundær effekt efter den første infusion. Når det administreres intravenøst til patienter med hæmofili, binder faktor VIII til von Willebrand-faktor i patientens blodomløb. Aktiv faktor VIII (VIla) fungerer som en kofaktor af aktiv faktor IX (IXa) for at fremskynde omdannelsen af faktor X til aktiv faktor X (Xa). Aktiv faktor X omdanner protrombin til thrombin. Thrombin omdanner derefter fibrinogen til fibrin, hvilket tillader dannelse af en blodprop. Hos patienter med hæmofili A forbliver ca. 2/3 til 3/4 af faktor VIII efter en injektion i kredsløbet. Faktor VIII aktivitetsniveau opnået i plasma skal være mellem 80% og 120% af den forudsagte faktor VIII aktivitet. Faktor VIII-aktivitet i plasma falder med en eksponentiel tofasefordeling. I den indledende fase forekommer fordelingen mellem intravaskulære og andre rum (kropsvæsker) med en plasmaeliminationshalveringstid på 3 til 6 timer. I den næste langsommere fase, som sandsynligvis afspejler faktor VIII-forbrug, er halveringstiden 8 til 18 timer med et gennemsnit på 15 timer. h.
Dosering
Intravenøst. Behandlingen skal initieres og overvåges af en læge med erfaring i behandling af blødningsforstyrrelser. Faktor VIII-mangel: Dosis og behandlingsvarighed afhænger af sværhedsgraden af faktor VIIII-mangel, placeringen og omfanget af blødningen og patientens kliniske tilstand. 1 IE Faktor VIII-aktiviteten svarer til mængden af faktor VIII i 1 ml normalt humant plasma. Administration af 1 IE faktor VIII / kg øger plasma faktor VIII aktivitet med ca. 1,7% - 2,5% af normal aktivitet, derfor beregnes den krævede dosis som følger: krævet antal enheder = kropsvægt (kg) x ønsket faktor VIII stigning (%) x 0,5 . Den mængde, der skal administreres, og administrationsfrekvensen skal altid afhænge af den individuelle kliniske effektivitet i et givet tilfælde (nogle gange, f.eks. I nærværelse af en lav inhibitor-titer, kan det være nødvendigt at bruge en dosis højere end den, der beregnes i henhold til formlen). Ved blødning og kirurgi bør faktor VIII-aktivitet over tid ikke falde under følgende aktivitetsniveauer (i% af normal eller IE / dL): tidlig blødning i led, muskler eller mund - det krævede faktor VIII-niveau er 20-40% af det normale og lægemidlet administreres hver 12-24 timer i mindst 1 dag, indtil blødningen er forbi (smertelindring) eller såret er helet; mere alvorlig blødning i led, muskler eller hæmatom - det krævede faktor VIII-niveau er 30-60% af det normale, og lægemiddelinfusioner gentages hver 12-24 timer i 3-4 dage eller længere, indtil smertelindring og akut funktionsnedsættelse er løst; livstruende blødning - det krævede faktor VIII-niveau er 60-100% af det normale, lægemiddelinfusioner gentages hver 8-24 timer, indtil truslen er løst; mindre operation (inklusive tandekstraktion) - det krævede faktor VIII-niveau er 30-60%, lægemidlet gives hver 24. time i mindst 1 dag, indtil såret er helet; større operation - krævede faktor VIII niveauer er 80-100% (før og efter operationen), infusioner gentages hver 8-24 timer, indtil såret er helet, fortsættes derefter i mindst 7 på hinanden følgende dage, idet faktor VIII niveauer holdes på 30-60 % (IU / dL). Respons på behandling varierer individuelt, og det er derfor vigtigt at overvåge faktor VIII-niveauer, især i større operationer. Ved den langsigtede profylakse af blødning ved svær hæmofili A er den sædvanlige dosis af faktor VIII 20-40 IE / kg. hver 2-3 dag kan kortere dosisintervaller eller højere doser være nødvendige hos yngre patienter. Von Willebrands sygdom. Det antages generelt, at indgivelsen af 1 IE af VWF: RCo / kg kropsvægt øger niveauet af VWF: RCo i kredsløbet med 2%. Målet med behandlingen er at opnå niveauet af VWF: RCo> 0,6 IE / ml (60%) og FVIII: C> 0,4 IE / ml (40%) i plasma. I de fleste tilfælde anbefales til hæmostase en dosis på 40-80 IE / kg kropsvægt af von Willebrand-faktor og 20-40 IE / kg kropsvægt på FVIII: C. Patienter med type 3 von Willebrand-sygdom, som muligvis har brug for højere doser for at opretholde tilstrækkelige niveauer af faktoren, kan kræve en indledende dosis von Willebrand-faktor på 80 IE / kg kropsvægt. Den valgte dosis skal administreres hver 12-24 timer. Dosering og behandlingsvarighed afhænger af patientens kliniske tilstand, placering og omfanget af blødningen og niveauet af både VWF: RCo og FVIII: C. Ved anvendelse af faktor VIII-præparater, der indeholder von Willebrand-faktor, skal den behandlende læge tage højde for muligheden for en overdreven stigning i FVIII: C-niveauer. For at undgå en overdreven stigning i FVIII: C-niveauer, dosisreduktion og / eller forlængelse af doseringsintervallet eller brugen af lægemidler, der indeholder VWF og mindre faktor VIII, bør overvejes efter 24-48 timers behandling. Kun begrænsede data fra kliniske forsøg med børn under 6 år er tilgængelige for ovenstående indikation - må ikke bruges. Hos børn i de ovennævnte indikationer udføres titrering til klinisk effektivitet også ved at beregne dosis i henhold til kropsvægt. Lægemidlet administreres som en langsom intravenøs injektion (indgivelseshastigheden bør ikke overstige 10 ml / min).
Indikationer
For at forhindre og kontrollere blødning hos patienter med hæmofili A (medfødt mangel på human koagulationsfaktor VIII). Forebyggelse og kontrol af blødning (inklusive kirurgisk blødning) hos patienter med von Willebrands sygdom (VWD), når behandling med desmopressin (DDAVP) er ineffektiv eller kontraindiceret. Præparatet kan anvendes til behandling af erhvervet mangel på human koagulationsfaktor VIII.
Kontraindikationer
Overfølsomhed over for det aktive stof eller over for et eller flere af hjælpestofferne.
Forholdsregler
Allergiske overfølsomhedsreaktioner er mulige. Patienterne bør informeres om de tidlige tegn på overfølsomhedsreaktioner, herunder udslæt, omfattende nældefeber, brysttæthed, hvæsen, hypotension, op til anafylaktisk chok. Hvis sådanne symptomer opstår, bør administrationen af lægemidlet afbrydes straks og passende behandling indledes efter behov. Patienter behandlet med human koagulationsfaktor VIII skal monitoreres omhyggeligt for udvikling af anti-faktor VIII-hæmmere ved klinisk observation og vurdering af laboratorietest. Risikoen for at udvikle hæmmere er korreleret med varigheden af eksponering for faktor VIII, hvor risikoen er størst i de første 20 eksponeringsdage. I sjældne tilfælde kan hæmmere også udvikle sig efter de første 100 eksponeringsdage. Tilbagevendende (lav titer) hæmmerdannelse er rapporteret hos patienter, der tidligere er blevet behandlet i mindst 100 dage med en hæmmende dannelse i historien efter skift fra et lægemiddel indeholdende faktor VIII. Derfor anbefales omhyggelig overvågning af patienter for forekomsten af hæmmere efter enhver skift til lægemiddel. Der er risiko for trombotiske komplikationer ved anvendelse af von Willebrand-faktor-lægemidler, især hos patienter med kendte kliniske risikofaktorer eller laboratoriefaktorer. Af denne grund er det nødvendigt at overvåge patienter i risiko for at opdage tidlige tegn på trombose. Derudover skal anbefalingerne i de nuværende retningslinjer for behandling og forebyggelse af venøs tromboembolisme følges. I behandlingsperioden med et lægemiddel, der indeholder faktor VIII og von Willebrand-faktor, bør lægen tage højde for, at langvarig behandling kan forårsage en overdreven stigning i FVIII: C-niveauer. Hos patienter behandlet med lægemidler, der indeholder faktor VIII og von Willebrand-faktor, skal FVIII: C-niveauer overvåges for at undgå vedvarende overskydende niveauer, der kan øge risikoen for trombotiske komplikationer. Patienter med von Willebrands sygdom, især type 3, kan udvikle neutraliserende antistoffer (hæmmere) mod von Willebrand-faktor. Hvis det forventede plasma-VWF: RCo-aktivitetsniveauer ikke opnås, eller blødning kontrolleres med passende doser, bør der undersøges for at kontrollere tilstedeværelsen af en von Willebrand-faktorhæmmer. Hos patienter med høje niveauer af inhibitorer er behandling med von Willbrand-faktor muligvis ikke effektiv, og andre terapeutiske muligheder bør overvejes. Hvis der kræves en central venelinie, kan der forekomme lokale infektioner, bakteriæmi og katetertrombose. Passende vaccination (mod hepatitis A- og B-vira) bør overvejes hos patienter, der regelmæssigt får plasma-afledt koagulationsfaktor VIII.
Uønsket aktivitet
Overfølsomhed eller allergiske reaktioner (angioødem, brændende og stikkende fornemmelse på injektionsstedet, kulderystelser, paroxysmal rødmen, generaliseret urticaria, hovedpine, udslæt, blodtryksfald, søvnighed, kvalme, rastløshed, takykardi, trykken i brystet, prikkende fornemmelse, opkastning, hvæsende vejrtrækning), hvilket i nogle tilfælde fører til svær anafylaksi med chok. Temperaturstigninger er sjældent observeret. Patienter med hæmofili A kan udvikle neutraliserende antistoffer (hæmmere) til faktor VIII. Når sådanne hæmmere udvikles, observeres et utilstrækkeligt klinisk respons på behandlingen. I meget sjældne tilfælde kan patienter med von Willebrand-sygdom, især type 3-sygdom, udvikle neutraliserende antistoffer (hæmmere) mod von Willebrand-faktor. Hvis sådanne hæmmere udvikles, observeres et utilstrækkeligt klinisk respons på behandlingen. Disse antistoffer kan være tæt forbundet med en anafylaktisk reaktion. Af denne grund bør patienter, der oplever anafylaktiske reaktioner, testes for tilstedeværelse af hæmmere. I sådanne tilfælde anbefales det at kontakte et specialiseret center for behandling af hæmostatiske lidelser. Der er risiko for trombotiske komplikationer, når lægemidlet anvendes til patienter med von Willebrand-sygdom med kendte kliniske eller laboratorierisikofaktorer.
Graviditet og amning
Præparatet bør kun anvendes under graviditet og amning, hvis det er klart nødvendigt (ingen erfaring med brugen af lægemidlet på grund af den sjældne forekomst af hæmofili A hos kvinder).
Interaktioner
Der er ingen kendte interaktioner mellem FVIII / VWF syndrom og andre præparater.
Præparatet indeholder stoffet: Faktor VIII, Faktor von Willebrand
Refunderet stof: NEJ






-wartoci-odywcze.jpg)




-po-zabiegach-kardiologicznych.jpg)