Spongiform encephalopatier (prionsygdomme) er de sygdomme, hvor patologiske former for prionproteiner er involveret i udviklingen. Vi ved mere og mere om prionsygdomme, men de vigtigste aspekter er stadig ukendte - medicin har i øjeblikket ikke midlerne til at helbrede patienter fra disse sygdomme.
Spongiforme encephalopatier eller prionsygdomme kan udvikles i løbet af livet, mens andre stammer fra arvelige genmutationer til stede fra fødslen. Inden for denne gruppe findes der flere enheder hos mennesker, eksempler er Creutzfeldt-Jakobs sygdom eller dødelig familiær søvnløshed.
Prionsygdomme har været meget mystiske i lang tid. I modsætning til andre patogener, såsom bakterier, vira eller svampe, indeholder de ikke nukleinsyre - prioner er kun lavet af proteiner. Teorien om prionsygdomme blev opdaget af S. Prusiner, denne opdagelse blev meget værdsat i det videnskabelige samfund - i 1997 blev forskeren tildelt Nobelprisen i medicin. Selv om der er gået relativt mange år siden prion-konceptet blev født, mener nogle forskere stadig, at det er ufuldstændigt og undersøger arten af disse forhold yderligere - nogle af de faktorer, der er ansvarlige for spongiforme encefalopatier, er nu blevet bekræftet.
Prionsygdomme: årsager
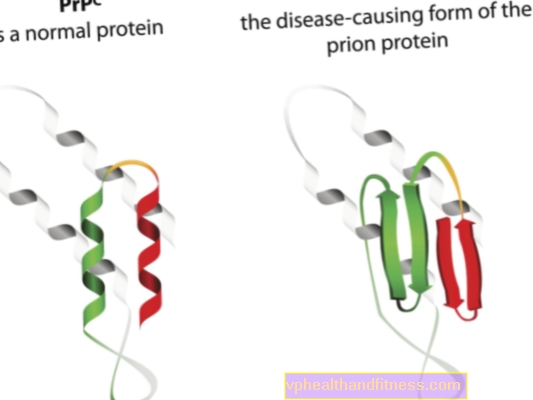
Etiologien af prionsygdomme er relateret til transformation af normale prionproteiner til patogene, patogene former. Prioner er proteinmolekyler, der findes i alles krop. Deres funktion er endnu ikke helt klar, men det er kendt, at prionproteiner under normale forhold ikke skader kroppen. Men når prioner ændrer deres struktur og bliver patogene partikler, udvikler en af flere spongiforme encefalopatier. Prioner, der naturligt forekommer i kroppen, kaldes PRPC, mens unormale former kaldes PRPSC. Sidstnævnte er et alvorligt problem ikke kun fordi de kan ophobes i nervevævet i form af aflejringer og generere dets skade, men også fordi de har evnen til at omdanne normale prioner til en misdannet form (enkelt sagt, PRPSC kan "inficere" normale proteiner med dets patogene potentiale).
Læs også: Huntingtons sygdom (Huntingtons chorea): årsager, symptomer, behandling Muskelskælv - årsager. Hvad betyder muskelskælv? Sygdomme, der dræber hurtigst: STØD, EBOLA, DAMN, ATTACK, NØD [GALE ...Dybest set er der 3 årsager til spongiform encefalopati:
- sporadisk (patogen mutation forekommer i somatiske celler, den forekommer i patientens liv),
- familie (som følge af byrden af mutationer arvet fra forældre),
- passeret (relateret til introduktion af patogene prioner i menneskekroppen, f.eks. gennem væksthormonpræparater, der er kontamineret med disse partikler eller hornhindetransplantation fra en person, der lider af en eller anden spongiform encefalopati).
Spongiform encefalopati: Creutzfeldt-Jakobs sygdom
Creutzfeldt-Jakobs sygdom (CJD) blev først beskrevet i de tidlige 1920'ere. Der er 4 typer af sygdommen:
- sporadisk CJD (den mest almindelige, tegner sig for op til 9/10 af alle CJD-sager)
- hjemby CJD
- bæltet CJD
- variant af CJD
Det kliniske billede i løbet af forskellige varianter af Creutzfeldt-Jakobs sygdom kan variere. De mest almindelige lidelser i løbet af denne gruppe af spongiforme encefalopatier er:
- demenslidelser (herunder progressiv forringelse af hukommelse, opmærksomhed og koncentration)
- myoklonus (ufrivillige bevægelser som pludselige muskelrykk)
- cerebellar dysfunktion (manifesteret for eksempel ved balanceforstyrrelser)
- sløret syn
- pyramidale og ekstrapyramidale symptomer
I løbet af CJD-varianter kan mentale lidelser (fx angst, deprimeret humør), smerte og andre ufrivillige bevægelser end de ovennævnte også forekomme.
Prognosen for Creutzfeldt-Jakobs sygdom er dårlig - for eksempel for patienter med sporadisk CJD tager det i gennemsnit fire til fem måneder fra sygdomssymptomers begyndelse til døden.
Spongiform encefalopati: Gerstmann-Straussler-Scheinker syndrom
Gerstmann-Straussler-Scheinker syndrom (GSS) kører normalt i familier og er forårsaget af en arvelig mutation i PRNP-genet. Det anses for at være den langsomt fremadgående spongiform encefalopati. GSS-teamet inkluderer:
- spinocerebellar ataksi
- dysartri
- demenslidelser
- synkeforstyrrelser
- nystagmus
- øget muskelspænding
Patienter diagnosticeret med GSS har en variabel tid, og hos nogle patienter opstår døden mere end 10 år efter indtræden.
Spongiform encefalopati: dødelig familiær søvnløshed
Dødelig familiær søvnløshed er en prionsygdom forårsaget af mutationer i PRNP-genet. Sygdommen er ekstremt sjælden og er indtil videre diagnosticeret i 28 familier over hele verden. I løbet af dødelig familiær søvnløshed er det første symptom manglende evne til at sove. Dette problem resulterer i angstlidelser og patienten oplever hallucinationer. Effekten af den konstante mangel på nattesøvn er dysfunktion i det autonome system (inklusive ændringer i hjertefunktion, svedtendens og fordøjelsessystemet), der er også et progressivt fald i kropsvægt. I mere avancerede stadier af dødelig familiær søvnløshed vises hormonelle forstyrrelser, og symptomer på demens opstår i løbet af sygdommen.
Prognosen for dødelig familiær søvnløshed, som for andre spongiforme encefalopatier, er dårlig: patienter dør normalt inden for tre år efter indtræden.
Spongiform encefalopati: prionopati med variabel følsomhed over for protease
Forekomsten af de diskuterede spongiforme encefalopatier er hovedsageligt relateret til mutationer i PRNP-genet. Disse mutationer vedrører imidlertid forskellige kodoner i dette gen, og der skelnes derfor adskillige forskellige prionsygdomme. En relativt nylig beskrevet (i 2008) enhed er prionopati med variabel modtagelighed for protease. Mennesker, der lider af denne sygdom, bærer mutationer i så mange som tre kodoner af PRNP-genet.
Ved prionopati med variabel følsomhed over for protease oplever patienterne:
- kognitiv svækkelse
- ekstrem sværhedsgrad af psykiatriske lidelser: de kan være eufori og agitation, men også betydelig apati
- dysartri
- afasi (sprogforstyrrelser)
Den gennemsnitlige varighed af sygdommen i denne prionopati er mindre end 4 år.
Spongiform encefalopati: kuru
Kuru betragtes nu som en sygdom, der praktisk talt ikke eksisterer længere - den blev fundet hos repræsentanter for stammer fra Papua Ny Guinea, der praktiserede kannibalistisk opførsel. Det dominerende symptom på denne spongiform encefalopati er progressiv cerebellær ataksi. Det kan ledsages af ufrivillige bevægelser (hovedsageligt i form af chorea, rysten og athetose) såvel som urin- og fæcesinkontinens. Patienter på kuru oplever også betydelige humørsvingninger, de udvikler primitive reflekser (fx sugning). Helt et karakteristisk problem i tilfælde af denne prionsygdom er tvungne anfald af gråd eller latter - på grund af sidstnævnte fænomen kaldes kuru undertiden som "latterdød".
Spongiform encefalopati: diagnose
Prionsygdomme kan mistænkes på baggrund af patientens symptomer. De er dog ret uspecifikke, da de også kan forekomme i løbet af en række andre sygdomme, der ikke er relateret til prioner. Af denne grund anvendes følgende også til diagnosen spongiform encefalopati:
- billeddannelsestest (fx magnetisk resonansbilleddannelse, som gør det muligt at detektere ændringer relateret til hjernens degeneration af prionproteiner),
- laboratorieundersøgelser (såsom vurdering af proteinkoncentrationer i cerebrospinalvæsken, f.eks. MAP-tau, S-100 eller 14-3-3 proteiner),
- genetiske tests (for at detektere tilstedeværelsen af mutationer hos patienten),
- immunhistokemiske tests (ved anvendelse af antistoffer mod prionproteiner).
Diagnosen kan også bekræftes ved obduktion af hjernen, hvor det er muligt at finde ændringer, der er karakteristiske for spongiforme encefalopatier. Disse kan være svampede læsioner, forskellig fordelt og med en anden struktur (afhængigt af den specifikke sygdomsenhed) amyloide plaques og neuronal defekter.
Spongiform encefalopati: behandling
Prionsygdomme er i øjeblikket uhelbredelige - på trods af adskillige undersøgelser, der har foregået i mange år, har medicin stadig ikke medicin, der kan bremse eller fuldstændigt hæmme deres fremskridt. Symptomatisk behandling anvendes til patienter med spongiform encefalopati, som har til formål at lindre intensiteten af symptomerne og forbedre deres livskvalitet så meget som muligt.
Arbejdet med behandling af spongiforme encefalopatier er dog stadig i gang. Forskere forsøger at bruge forskellige metoder - det første eksempel er genterapi. De ville påvirke nukleinsyrer og mutationerne i deres struktur - formålet med at anvende genterapi ville være at neutralisere fejl i den genetiske kode. En anden tilgang er grundlaget for immunterapi - der arbejdes på at skabe antistoffer, hvis rolle ville være at eliminere patogene prioner. En anden metode, der ser potentialet til at bekæmpe spongiforme encephalopatier, er behandling med anvendelse af syntetiserede proteinmolekyler, som, når de først er introduceret i patientens krop, vil neutralisere patologiske proteiner.
Anbefalet artikel:
Encefalopatier - årsager, typer og symptomer